Wingert 2026 — ferret auditory cortex encoding subspace
![]()
This notebook is a visual smoke test of the deepSTRF loader for the Wingert et al. 2026 (Nat Neurosci) ferret AC dataset — single-unit Kilosort-sorted spikes from 67 high-density silicon-probe + Neuropixels recordings spanning A1, PEG (and ancillary AC / HC), in 4 ferrets passively listening to natural-sound segments.
We cover:
the two stim-duration cohorts (20 s vs 22 s sessions) and the est / val subset semantics;
per-cell metadata: area, layer, depth, narrow / regular spike-width classes, the published
goodpredflag;the block-diagonal layout that pools two recording sessions — cross-session
(stim, cell)pairs are NaN sentinels;how the two-probe SLJ032a recording is exposed as two siteids that share one .tgz / one stim set.
Setup — Google Colab
The cell below installs deepSTRF from source on Colab. On a local install (pip install -e .) it’s a no-op.
Note on data. The Zenodo archive’s recordings.zip is ~4.35 GB; download=True fetches it on demand and skips the much larger wav.zip and models.zip that the loader doesn’t use. On a workstation with the data already unpacked, point WINGERT2026_DATA at the directory holding recordings/ and cell_list.csv.
[1]:
import sys
if 'google.colab' in sys.modules:
!pip install -q git+https://github.com/urancon/deepSTRF.git
print("deepSTRF installed from GitHub.")
else:
print("Local environment — assuming deepSTRF is already importable.")
Local environment — assuming deepSTRF is already importable.
Imports + paths
[2]:
%matplotlib inline
import os
import warnings
from collections import Counter
import numpy as np
import matplotlib.pyplot as plt
import torch
from deepSTRF.datasets.audio import Wingert2026Dataset
from deepSTRF.utils import plot_stim_with_response
# Where the Zenodo dump lives. None → platformdirs cache + download=True
# (4.35 GB recordings.zip + 5 MB cell_list.csv).
DATA = os.environ.get("WINGERT2026_DATA", None)
DOWNLOAD = False # flip to True on first run if the data isn't local
# Silence the (expected) PRN018b duplicate-drop UserWarning emitted on
# the first .tgz scan — see README §"Note on the PRN018a / PRN018b
# duplicate".
warnings.filterwarnings("ignore", category=UserWarning,
module=r"deepSTRF\.datasets\.audio\.wingert2026")
/home/ulysse/miniconda3/envs/deepstrf_dev/lib/python3.11/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
1. Per-cell metadata — A1 + PEG headline cohort (enumerate-only)
The _enumerate_only=True path reads only cell_list.csv (~5 MB) and populates nrn_meta / N_neurons without touching the per-site .tgz archives. Useful for fast metadata exploration and sanity checks before the heavy load. The area= and site= filters apply at this stage.
[3]:
ds_meta = Wingert2026Dataset(
path=DATA, download=DOWNLOAD, area=["A1", "PEG"], _enumerate_only=True,
)
print(f"A1 + PEG cohort: N = {ds_meta.N_neurons} ({ds_meta.N_neurons == 2128 + 746})")
# breakdown by area
by_area = Counter(m["area"] for m in ds_meta.nrn_meta)
print(f" by area: {dict(by_area)}")
# layer breakdown (column is a string like '1-3', '4', '56')
by_layer = Counter(m["layer"] for m in ds_meta.nrn_meta)
print(f" by layer: {dict(by_layer)}")
# narrow vs broad spike width
by_narrow = Counter(m["narrow"] for m in ds_meta.nrn_meta)
print(f" by narrow: {dict(by_narrow)}")
# published auditory-responsive flag
goodpred_frac = sum(1 for m in ds_meta.nrn_meta if m["goodpred"]) / len(ds_meta.nrn_meta)
print(f" goodpred=True fraction: {goodpred_frac:.1%}")
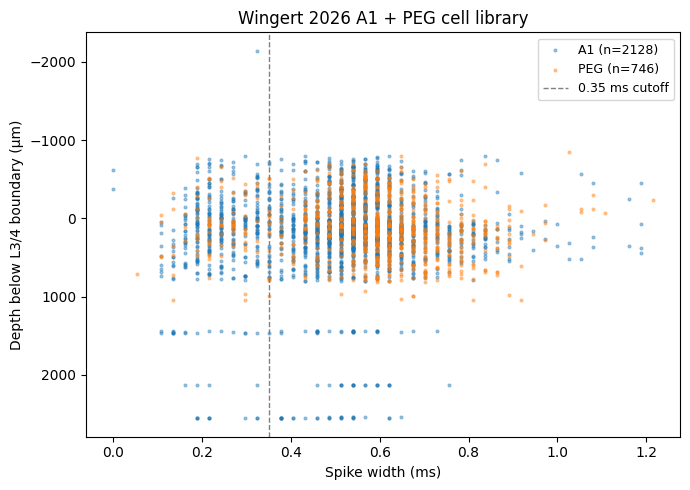
A1 + PEG cohort: N = 2874 (True)
by area: {'PEG': 746, 'A1': 2128}
by layer: {'56': 1084, '44': 744, '13': 1046}
by narrow: {False: 2397, True: 477}
goodpred=True fraction: 81.3%
Cell-depth + spike-width distribution
The David lab classifies cells as Regular vs Narrow spiking based on spike-waveform width (cutoff 0.35 ms for 64-channel silicon probes, 0.375 ms for Neuropixels). Narrow-spiking cells are putative inhibitory interneurons.
[4]:
# Depth + spike-width scatter, coloured by area
fig, ax = plt.subplots(figsize=(7, 5))
for area, color in [("A1", "tab:blue"), ("PEG", "tab:orange")]:
sub = [m for m in ds_meta.nrn_meta
if m["area"] == area and m["depth"] is not None and m["sw"] is not None]
ax.scatter(
[m["sw"] for m in sub], [m["depth"] for m in sub],
s=4, alpha=0.4, color=color, label=f"{area} (n={len(sub)})",
)
ax.axvline(0.35, color="grey", lw=1, linestyle="--", label="0.35 ms cutoff")
ax.set_xlabel("Spike width (ms)")
ax.set_ylabel("Depth below L3/4 boundary (μm)")
ax.invert_yaxis() # superficial layers up
ax.set_title("Wingert 2026 A1 + PEG cell library")
ax.legend(loc="best", fontsize=9)
plt.tight_layout(); plt.show()
2. Load one small site (CLT027c, 20 cells in A1)
A single-site Wingert2026Dataset instance is cheap (~7 s on a laptop, <500 MB peak RSS). Because every cell saw every stim, the response grid has no NaN sentinels at this scale.
[5]:
ds = Wingert2026Dataset(path=DATA, download=DOWNLOAD, site="CLT027c")
print(f"site='CLT027c' N={ds.N_neurons} S={len(ds.stims)} F={ds.F} dt={ds.dt} ms")
# Stim subsets (file-name prefix: STIM_00* = val, STIM_seq* = est)
sub_counts = Counter(m["subset"] for m in ds.stim_meta)
print(f" subset counts: {dict(sub_counts)}")
# This site is in the 22 s cohort (T=2200 = 1 s pre + 20 s sound + 1 s post)
print(f" stim shapes: {sorted({tuple(s.shape) for s in ds.stims})}")
# val stim R: this site presented one val stim with R=1, the other with R=2
for i, m in enumerate(ds.stim_meta):
if m["subset"] == "val":
print(f" val stim {m['name']!r} → R={tuple(ds.responses[i][0].shape)[0]}")
Wingert2026 sites: 100%|██████████| 1/1 [00:00<00:00, 11.79it/s]
site='CLT027c' N=20 S=11 F=32 dt=10.0 ms
subset counts: {'val': 2, 'est': 9}
stim shapes: [(1, 32, 2200)]
val stim 'STIM_00seq1.wav' → R=1
val stim 'STIM_00seq2.wav' → R=2
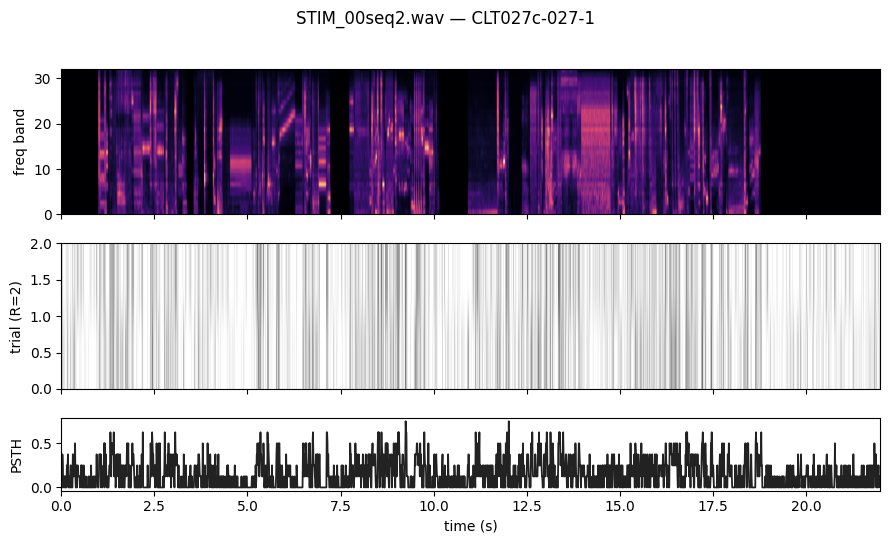
One stim, one cell — PSTH + spectrogram
plot_stim_with_response ships in deepSTRF.utils and accepts either NumPy arrays or torch tensors. We pick the val stim with the highest R count (most informative PSTH) and overlay all repeats from the spike-train cell with the highest mean firing rate.
[6]:
# pick the val stim with the highest R
val_indices = [i for i, m in enumerate(ds.stim_meta) if m["subset"] == "val"]
s_idx = max(val_indices, key=lambda i: ds.responses[i][0].shape[0])
spec = ds.stims[s_idx][0] # (F, T)
print(f"showing stim {ds.stim_meta[s_idx]['name']!r} with R={ds.responses[s_idx][0].shape[0]}")
# pick the most-active cell across this stim
mean_rates = [ds.responses[s_idx][n].mean().item() for n in range(ds.N_neurons)]
n_idx = int(np.argmax(mean_rates))
cell_meta = ds.nrn_meta[n_idx]
print(f"showing cell {cell_meta['cell_id']!r} (area={cell_meta['area']}, "
f"layer={cell_meta['layer']}, narrow={cell_meta['narrow']})")
reps = ds.responses[s_idx][n_idx] # (R, T)
# plot_stim_with_response returns (Figure, axes). It auto-shows the
# raster panel when R > 1 and adds the PSTH overlay.
fig, _ = plot_stim_with_response(
spec, reps, dt_ms=ds.dt,
title=f"{ds.stim_meta[s_idx]['name']} — {cell_meta['cell_id']}",
)
plt.show()
showing stim 'STIM_00seq2.wav' with R=2
showing cell 'CLT027c-027-1' (area=A1, layer=56, narrow=True)
3. Block-diagonal layout — load two sites at once
Loading more than one site is where the deepSTRF sparse-coverage paradigm earns its keep. Each cell only has real data for the ~100 stimuli its session presented; for every other session’s stimuli, the loader emits a (1, 1) NaN sentinel — and crucially all sentinels share the same underlying tensor, so the in-memory overhead is one pointer per slot, not a fresh tensor.
[7]:
ds2 = Wingert2026Dataset(path=DATA, download=DOWNLOAD, site=["CLT027c", "CLT028c"])
print(f"two-site load: N={ds2.N_neurons} S={len(ds2.stim_meta)} (sessions: "
f"{sorted(set(m['session'] for m in ds2.stim_meta))})")
# count real-data slots per session
by_session_real = Counter()
by_session_sentinel = Counter()
for s, smeta in enumerate(ds2.stim_meta):
for n, nmeta in enumerate(ds2.nrn_meta):
if ds2.responses[s][n].numel() == 1:
by_session_sentinel[(smeta["session"], nmeta["session"])] += 1
else:
by_session_real[(smeta["session"], nmeta["session"])] += 1
print(f"\nreal-data (stim_session × cell_session) counts:")
for key, n in sorted(by_session_real.items()):
print(f" {key}: {n}")
print(f"\nsentinel counts:")
for key, n in sorted(by_session_sentinel.items()):
print(f" {key}: {n}")
# Memory regression check: all NaN sentinels share one id
sentinel_ids = {id(t) for row in ds2.responses for t in row if t.numel() == 1}
print(f"\nunique sentinel object ids: {len(sentinel_ids)} (expect 1)")
Wingert2026 sites: 100%|██████████| 2/2 [00:01<00:00, 1.52it/s]
two-site load: N=77 S=117 (sessions: ['CLT027c', 'CLT028c'])
real-data (stim_session × cell_session) counts:
('CLT027c', 'CLT027c'): 220
('CLT028c', 'CLT028c'): 6042
sentinel counts:
('CLT027c', 'CLT028c'): 627
('CLT028c', 'CLT027c'): 2120
unique sentinel object ids: 1 (expect 1)
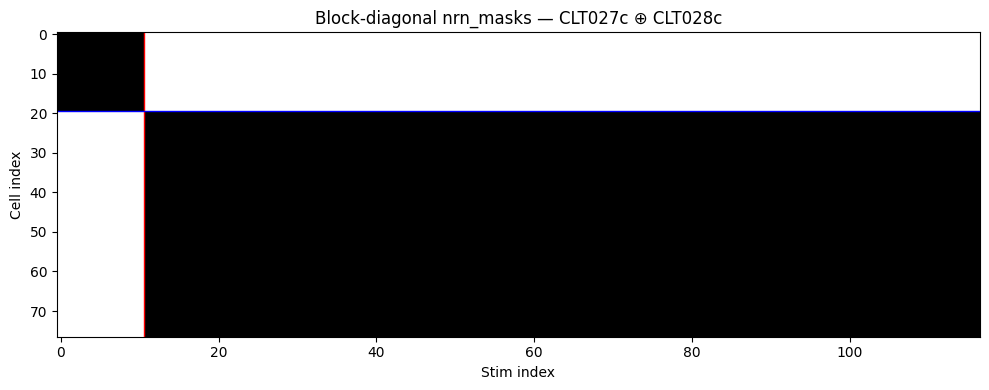
Visualising the block-diagonal coverage
The nrn_masks (S, N) bool tensor (auto-derived from NaN sentinels) makes the layout immediately visible. The grid is clean block-diagonal: session A’s stims have real data only for session A’s cells.
[8]:
m = ds2.nrn_masks.numpy() # (S, N) bool
fig, ax = plt.subplots(figsize=(10, 4))
ax.imshow(m.T, aspect="auto", cmap="Greys", interpolation="nearest")
# Session boundary lines
sess_seen = []
for s, smeta in enumerate(ds2.stim_meta):
sess = smeta["session"]
if sess_seen and sess_seen[-1] != sess:
ax.axvline(s - 0.5, color="red", lw=1)
sess_seen.append(sess)
cell_seen = []
for n, nmeta in enumerate(ds2.nrn_meta):
sess = nmeta["session"]
if cell_seen and cell_seen[-1] != sess:
ax.axhline(n - 0.5, color="blue", lw=1)
cell_seen.append(sess)
ax.set_xlabel("Stim index")
ax.set_ylabel("Cell index")
ax.set_title("Block-diagonal nrn_masks — CLT027c ⊕ CLT028c")
plt.tight_layout(); plt.show()
4. SLJ032a — one .tgz, two probes, one stim set
The SLJ032a recording used two simultaneously-inserted probes, A and B. cell_list.csv exposes them as separate siteids ('SLJ032a' for probe A’s 76 cells, 'SLJ032a-B' for probe B’s 47 cells), but they share one .tgz / one stim set. Loading both probes gives 123 cells and one stim list (no duplication).
[9]:
ds_slj = Wingert2026Dataset(
path=DATA, download=DOWNLOAD, site=["SLJ032a", "SLJ032a-B"],
)
print(f"SLJ032a both probes: N={ds_slj.N_neurons} S={len(ds_slj.stim_meta)} "
f"sessions={sorted(set(m['session'] for m in ds_slj.stim_meta))}")
# Site breakdown
by_site = Counter(m["site"] for m in ds_slj.nrn_meta)
print(f" by site (per-probe): {dict(by_site)}")
# Cell-id format breakdown — probe A and probe B use distinct 4-segment prefixes
by_prefix = Counter("-".join(m["cell_id"].split("-")[:2]) for m in ds_slj.nrn_meta)
print(f" by cell-id prefix: {dict(by_prefix)}")
Wingert2026 sites: 100%|██████████| 1/1 [00:00<00:00, 1.31it/s]
SLJ032a both probes: N=123 S=56 sessions=['SLJ032a']
by site (per-probe): {'SLJ032a': 76, 'SLJ032a-B': 47}
by cell-id prefix: {'SLJ032a-A': 76, 'SLJ032a-B': 47}
5. The two duration cohorts
47 sites at T=2000 bins (20 s sound, no silence flanks) coexist with 21 sites at T=2200 bins (1 s pre + 20 s sound + 1 s post silence). The deepSTRF data paradigm handles ragged T natively — collate zero-pads on the right; models that expect a fixed T can use select_stims_by_predicate to filter to one cohort.
[10]:
# Enumerate cohorts from the local Phase 2 scan (no full load needed)
ds_a = Wingert2026Dataset(path=DATA, download=DOWNLOAD, area="A1", _enumerate_only=True)
sites = sorted(set(m["session"] for m in ds_a.nrn_meta))
print(f"A1 has cells in {len(sites)} unique recording sessions.")
print("(Stim-duration cohort is property of the .tgz itself; use a load to confirm.)")
A1 has cells in 50 unique recording sessions.
(Stim-duration cohort is property of the .tgz itself; use a load to confirm.)
What’s next
Linear / CNN baselines — the
Fitterfromdocs/_source/md/fitter.mdports to Wingert unchanged; usesubset='est'for train/val andsubset='val'for held-out evaluation. Note the per-site R variability for val stims (5–30) when reporting cc_norm.Cross-area pooling —
concat_neural_datasets([a1, peg])builds a 2 874-cell joint dataset for unified-model fitting. Like NAT4, the joint dataset is block-diagonal across areas as well as sessions.The published encoding subspace — the paper’s CNN / LN / subspace fits live in
models.zipon Zenodo. They are NEMS-format and not loaded by deepSTRF; re-fitting against the deepSTRF model zoo (Linear, StateNet, Transformer) is the natural next step.